LiCoRNA: aLignment of Complex RNAs
A tool for structure sequence alignment
Our News
17 Apr 2017
LiCoRNA v1.0 is released
The first version of LiCoRNA contains three main programs: alignerDP, alignerSB, alignerMEA. You could download here
Support






Tutorials
Quick start
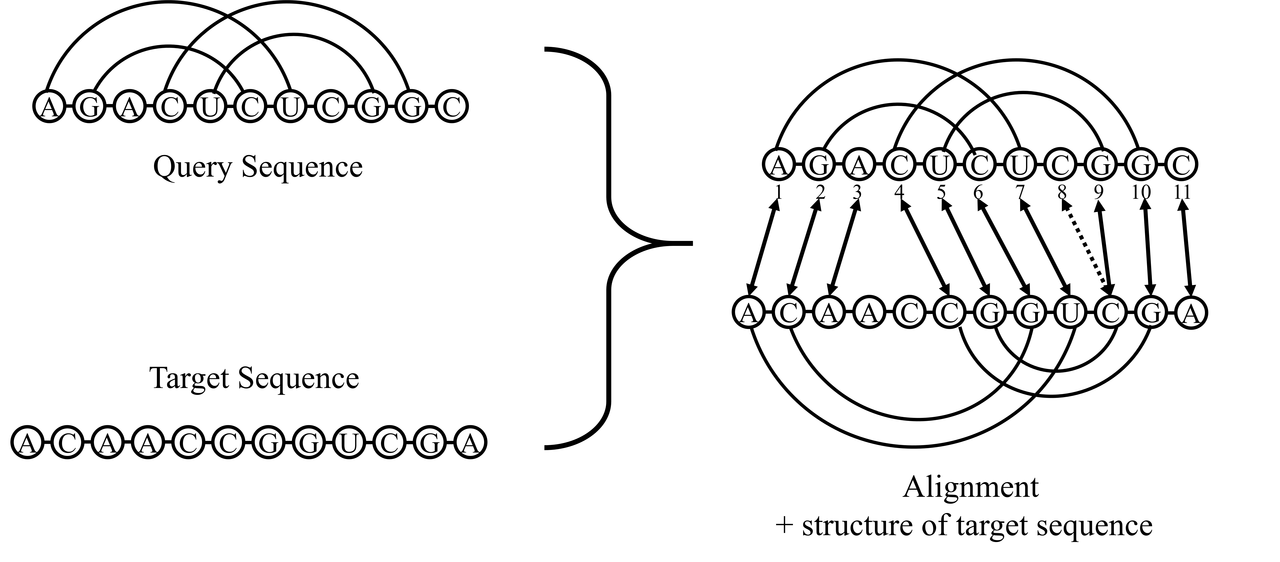
LiCoRNA is a software for structure sequence alignment for two RNAs in the presence of arbitrary pseudoknots. That is to say, when the structure of one ncRNA in the family has been determined by experiment, the other members of the family can be modeled based on their alignment to the known structure.
Input of alignerDP, alignerSB and alignerMEA
The input for programs alignerDP, alignerSB and alignerMEA needs two file: one file contains the query sequence and the other file contains the target sequence. Several points should be considered about the input file:
- Accepted symbols for sequence are: 'A', 'C', 'G' and 'U'
- Accepted symbols for structure are: '.', '[' and ']', '(' and ')', '<' and '>', '{' and '}'. If the four symbols are still not enough for you, then the symbols with 'A' and 'a', 'B' and 'b', 'C' and 'c' ... 'Z' and 'z' are also available
- Support any kind of pseudoknots
- support Watson-Crick base pairs and G-U base pair. DO NOT support non-Watson–Crick base pairs
Here is an example for the query sequence file
| A | G | A | C | U | C | U | C | G | G | C |
| < | < | . | ( | ( | > | > | . | ) | ) | . |
Here is an example for the target sequence file
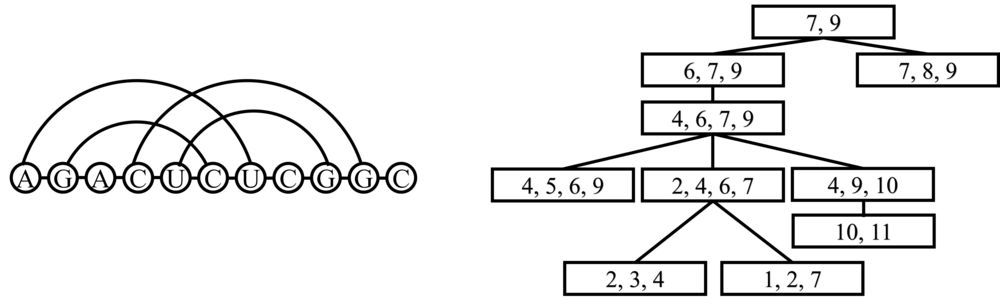
Tree Decomposition of query sequence
The RNA query sequence could be treated as RNA graph interaction structure (RNA graph). Tree decomposition give a way to map a graph to a tree. Program RNAML2dgf transfer the RNA sequence sequence file to dgf file which is needed for the tree decomposition. The program TreeDecomposer writen in Java is used for tree decomposition of query sequence. Below list the commands and the tree decomposition of an example query sequence is shown below.
alignerDP
alignerDP offers an methods to computer the optimal alignment between query sequence and target sequence.
OPTION
-i input path of RNA folded sequence.
-d tree decomposition of the query sequence in dot format.
-s intput path of the target sequence.
-c restrict the maximum insertion length
-n number of suboptimal alignment, the default value is 5.
In our case, the command could be:
alignerSB
alignerSB offers an methods to generate a representative sample of alignment using stochastic sampling.
OPTION
-i input path of RNA folded sequence.
-d tree decomposition of the query sequence in dot format.
-s intput path of the target sequence.
-c restrict the maximum insertion length
-n number of sampling aignments, the default value is 100.
-t temperature.
In our case, the command could be:
alignerMEA
alignerMEA offers an methods to computer compute an alignment with maximum expected accuracy over all alignments that are sampled by stochastic sampling.
OPTION
-i input path of RNA folded sequence.
-d tree decomposition of the query sequence in dot format.
-s intput path of the target sequence.
-c restrict the maximum insertion length
-t temperature.
In our case, the command could be:
Visualization
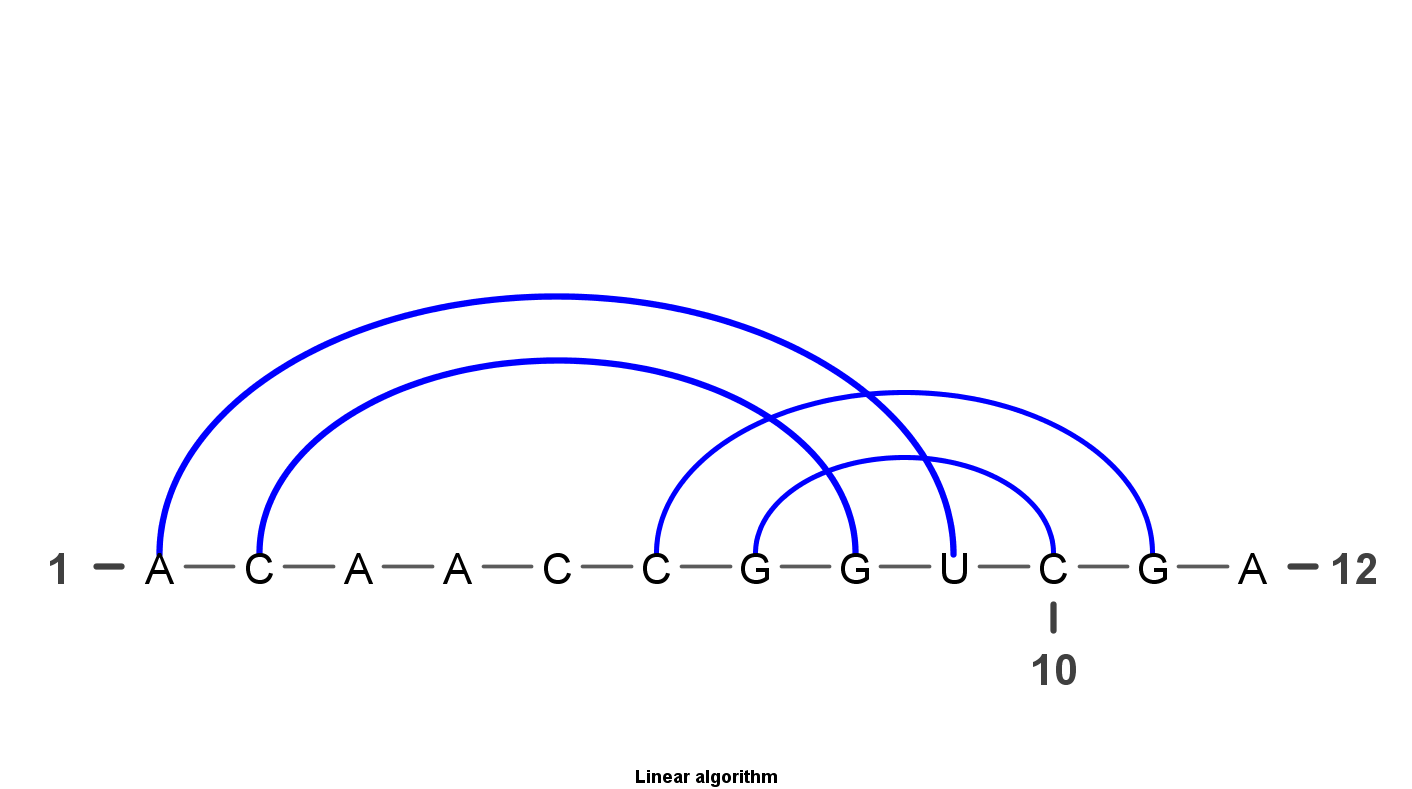
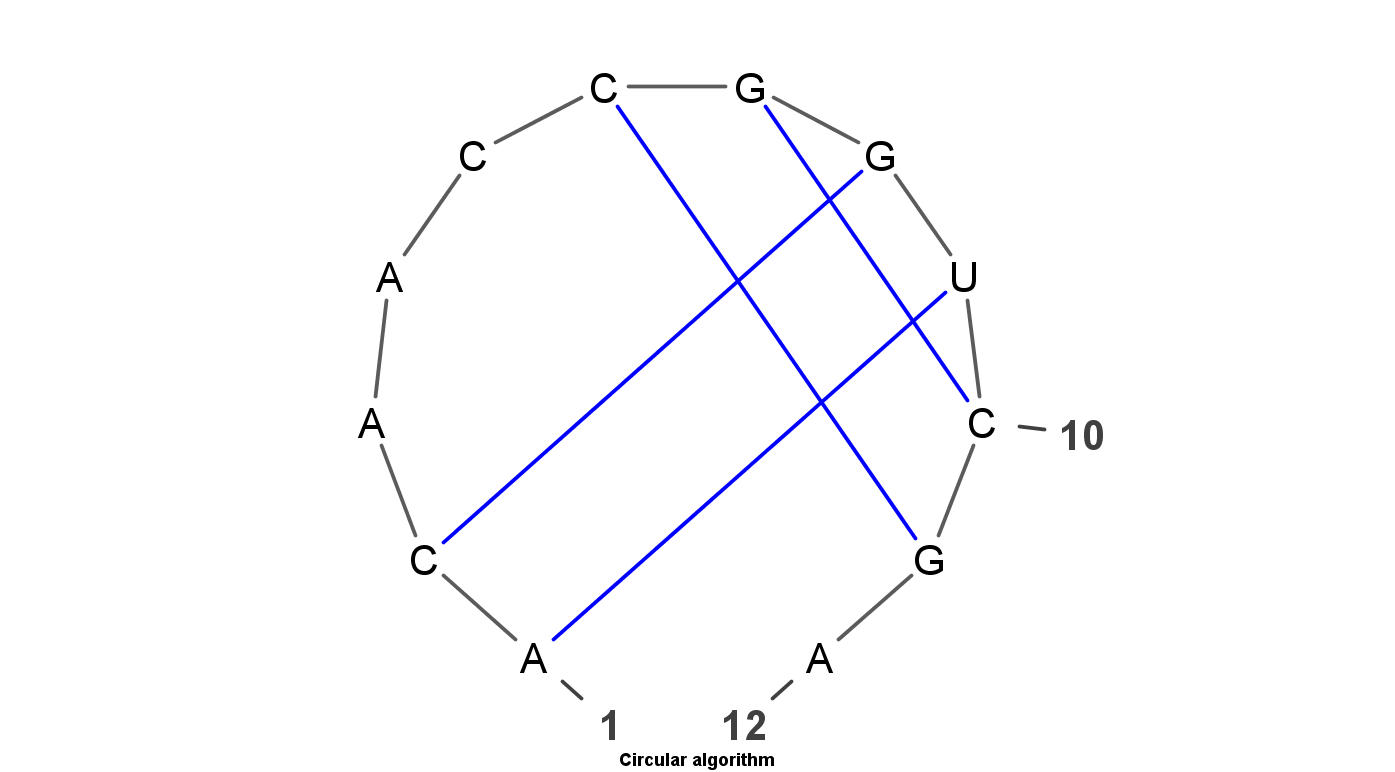
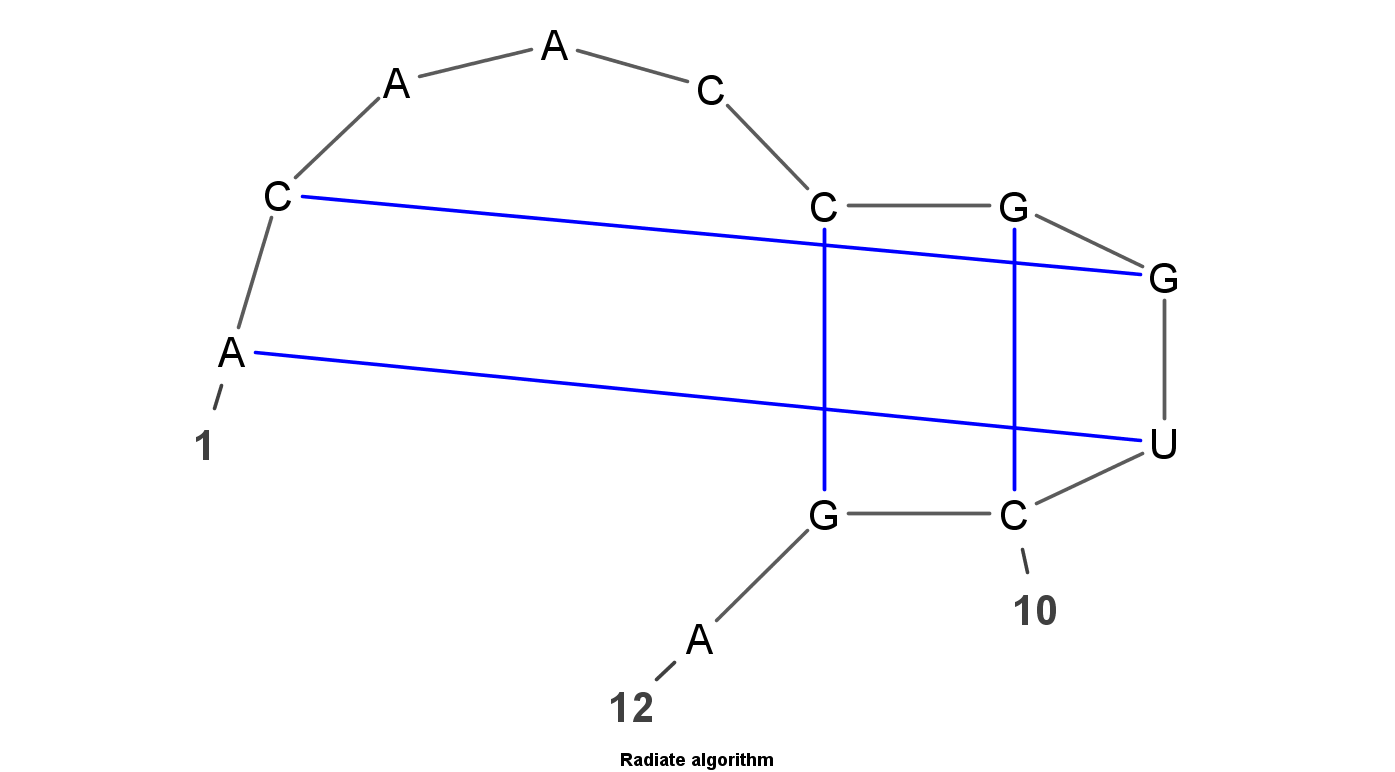
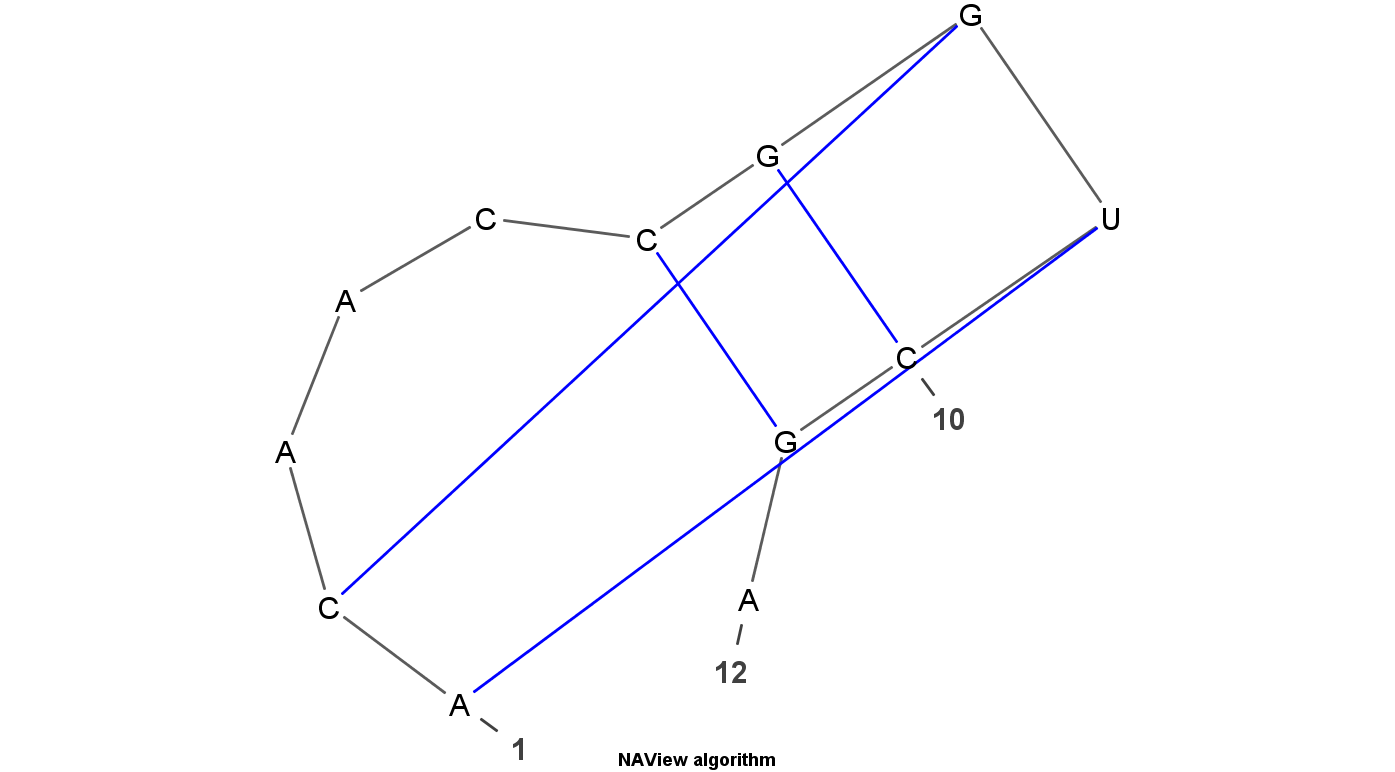
VARNA is Java lightweight Applet dedicated to drawing the secondary structure of RNA. It is developed by Yann Ponty . It mainly have four algorithm to display the RNA structure. Top left uses the linear algorithm and top right uses circular algorithm. Down left uses the Radite algorithm and down right uses the NAView algorithm .